Der Prozesstechnologie-Spezialist GEA stellte auf der Weltleitmesse CPHI/PMEC India eine Innovation für die pharmazeutische Fertigungsindustrie vor: die Hochleistungs-Rotationstablettenpresse Performa D....
Auch in Produktions- und Laborumgebungen mit hohen Hygiene- und Sicherheitsanforderungen setzen Unternehmen zunehmend auf die Flexibilität mobiler Endgeräte. Mit der...
Der Prozesstechnologie-Spezialist GEA stellte auf der Weltleitmesse CPHI/PMEC India eine Innovation für die pharmazeutische Fertigungsindustrie vor: die Hochleistungs-Rotationstablettenpresse Performa D....
Die Rückverlagerung pharmazeutischer Produktionskapazitäten nach Europa wird für viele Unternehmen zunehmend zu einem strategisch wichtigen Erfolgsfaktor. Saltigo, die Exklusivsynthese-Tochter von...
Der Prozesstechnologie-Spezialist GEA stellte auf der Weltleitmesse CPHI/PMEC India eine Innovation für die pharmazeutische Fertigungsindustrie vor: die Hochleistungs-Rotationstablettenpresse Performa D....
Auch in Produktions- und Laborumgebungen mit hohen Hygiene- und Sicherheitsanforderungen setzen Unternehmen zunehmend auf die Flexibilität mobiler Endgeräte. Mit der...
Der Prozesstechnologie-Spezialist GEA stellte auf der Weltleitmesse CPHI/PMEC India eine Innovation für die pharmazeutische Fertigungsindustrie vor: die Hochleistungs-Rotationstablettenpresse Performa D....
Die Rückverlagerung pharmazeutischer Produktionskapazitäten nach Europa wird für viele Unternehmen zunehmend zu einem strategisch wichtigen Erfolgsfaktor. Saltigo, die Exklusivsynthese-Tochter von...
Angesichts der globalen Marktgröße für Kombinationsprodukte, die bis 2024 voraussichtlich 177,7 Mrd. USD erreichen wird , und der zunehmenden technologischen Innovation auf dem Markt für Medizinprodukte, ist es von entscheidender Bedeutung, sicherzustellen, dass die Regulierung für Kombinationsprodukte den sich entwickelnden Markt widerspiegelt und die Sicherheit und Wirksamkeit erhöht. Ein weiteres wichtiges Thema, das ebenfalls in diesem Artikel behandelt wird, ist die neue EU-Verordnung über In-vitro-Diagnostika (IVDR). Diese enthält Anforderungen für die Regelung der therapiebegleitenden Diagnostika.
Demgemäß führen die EU-Medizinprodukteverordnung (MDR) und die neue EU-Verordnung über In-vitro-Diagnostika (IVDR) wesentliche Veränderungen in die Regulierungslandschaft ein. Von diesen Veränderungen sind neben Medizinprodukteherstellern auch Pharmaunternehmen betroffen, die Kombinationsprodukte oder Begleitdiagnostika herstellen. Die Fristen für die Einhaltung der Vorschriften rücken immer näher. Medizinproduktehersteller und Pharmaunternehmen haben Zeit bis Mai 2020 beziehungsweise Mai 2022 diese einzuhalten.
Hersteller werden dazu aufgerufen, die verschärften Vorschriften aufmerksam aufzunehmen und umzusetzen, um ihren Zugang zum EU-Markt weiterhin zu gewährleisten. Da die Fristen für die Einhaltung der Vorschriften immer näher rücken, ist ein klares Verständnis der neuen Anforderungen und ein proaktiver Ansatz von äußerster Dringlichkeit. Die neuen Verordnungen decken viele Regelungslücken in der Branche auf. Untersuchungen haben besorgniserregende Anzeichen ergeben: Im Juni 2018 gaben 78 Prozent der 220 befragten Medizinproduktehersteller an, dass sie die EU-MDR nicht ausreichend verstehen. Es besteht eine sehr hohe Wahrscheinlichkeit, dass dies immer noch der Fall ist. Die Herausforderung im Bereich der Kombinationsprodukte besteht darin, dass Pharmaunternehmen zum ersten Mal einer Prüfung durch eine Benannte Stelle unterzogen werden müssen und nicht mit den Prozeduren vertraut sind. Fehlendes Wissen über die Medizinprodukteverordnung und die Kommunikationsabläufe mit Benannten Stellen ist zeit- und kostenaufwendig.
In diesem Artikel wird dargelegt, was Hersteller von Kombinationsprodukten wissen müssen, um auf die Einhaltung der EU-MDR hinzuarbeiten, welche Herausforderungen sie sich stellen und wie sie diese bewältigen können.
Kombinationsprodukte
Kombinationsprodukte sind therapeutische und diagnostische Produkte, die Arzneimittel, Geräte und/oder aktive biologische Wirkstoffe miteinander verbinden. Sie können aus einer beliebigen Kombination eines Arzneimittels, eines Geräts oder eines aktiven biologischen Wirkstoffs bestehen. In der Europäischen Union werden Kombinationsprodukte derzeit wie folgend reguliert: entweder als Arzneimittel, wenn die Arzneimittelkomponente die hauptsächliche Funktion und keine unterstützende Funktion im Rahmen des Produkts besitzt; oder als Medizinprodukt, wenn die Arzneimittelkomponente einen integralen, aber nebensächlichen Bestandteil trägt.
In der neuen EU-MDR wird der Begriff „Kombinationsprodukt“ nicht verwendet. Dies ist ein Begriff, der von der US-amerikanischen Food and Drug Administration (FDA) verwendet wird.
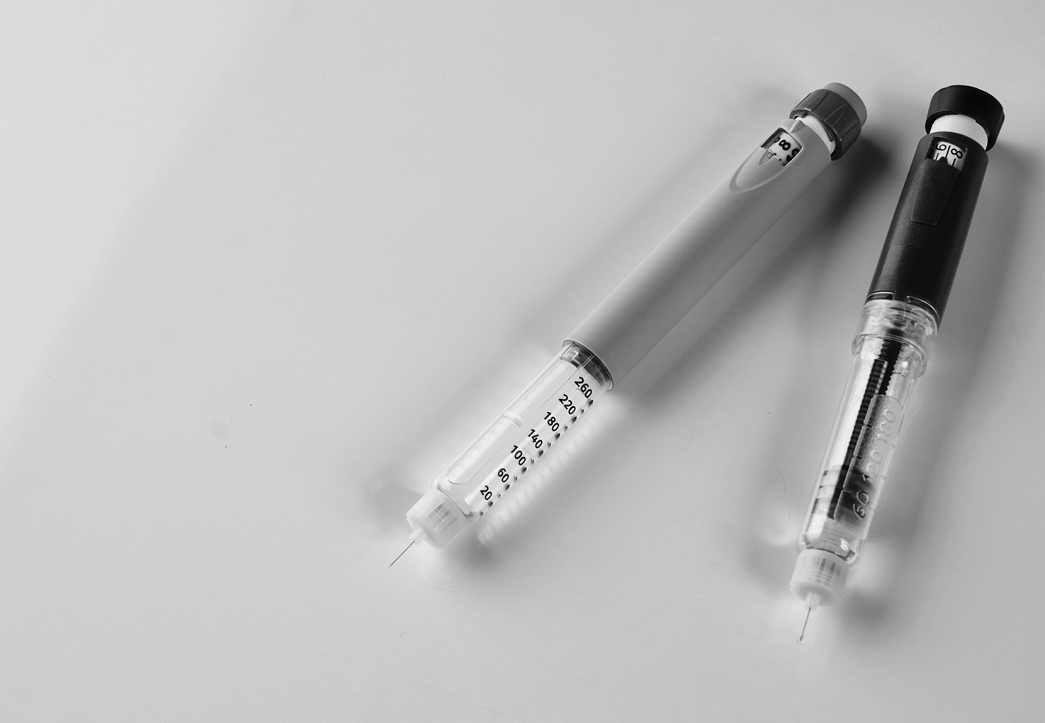
Etwa jedes vierte zentral zugelassene Arzneimittel enthält ein Medizinprodukt als Komponente, wobei die Mehrzahl davon ein integrales Gerät umfasst. Hierzu gehören beispielsweise Fertigspritzen und ‑stifte, Pflaster für die transdermale Arzneimittelabgabe und Fertiginhalatoren.
Regulatorische Hintergründe
Vor dem Eintreten des MDR, fielen Kombinationsprodukte unter zwei verschiedenen Richtlinien. Kombinationsprodukte, die als Medizinprodukte reguliert wurden, fielen unter dem Vorgänger des EU-MDR, die Medizinprodukterichtlinie (MDD), während Produkte, die als Arzneimittel reguliert wurden, unter die Arzneimittelrichtlinie (Medicinal Product Directive — MPD) fielen. Weder die MPD noch die MDD lagen klare regulatorische Anforderungen für das Medizinprodukt-Teil dar und gingen auch nicht näher darauf ein, wie Bewertungen durchgeführt werden sollten oder welche Unterlagen zum Nachweis der Konformität erforderlich sind. In einigen Fällen waren sich Pharmahersteller der MDD-Anforderungen möglicherweise nicht bewusst, zumal sie häufig nicht mit der medizinischen Geräteverordnung vertraut sind und möglicherweise nicht über ausreichendes internes Fachwissen über die Gerätekonformität verfügen. Mit dem technologischen Fortschritt in der Medizingeräte-Branche, wird das Thema zunehmend dringlicher. Zum Beispiel können Sensoren in „digitalen Pillen“ nachweisen, wann und in welcher Dosis Patienten Medikamente einnehmen. Da der Sensor geschluckt wird, können kleine Fehler lebensbedrohliche Folgen haben. Bisher fehlte es an regulatorischer Harmonisierung, da der Prüfungsgrad durch zuständige Behörden unterschiedlich war.
Artikel 117
In Artikel 117 des EU-MDR werden Regelungslücken ans Licht gebracht. Darin wird festgelegt, dass das von Pharmaunternehmen eingereichte Zulassungsdossier den Nachweis der Konformität des Produkts enthalten muss. Es gab bereits behördliche Kontrollen, die jedoch jetzt viel strenger sind. Somit wird sichergestellt, dass Gerätekomponente nicht der behördlichen Kontrolle entgehen.
Der Konformitätsnachweis kann auf zwei Arten erbracht werden:
Ergebnisse der Bewertung der Konformität des Medizinprodukte-Teils mit den relevanten allgemeinen Sicherheits- und Leistungsanforderungen von Anhang I des MDR, die in der EU-Konformitätserklärung des Herstellers enthalten sind;
oder die entsprechende Bescheinigung ausgestellt durch eine Benannte Stelle, mit dem der Hersteller das Medizinprodukt mit einer CE-Kennzeichnung versehen kann.
Auf welche Änderungen müssen sich Pharmaunternehmen gefasst machen?
Die größte Herausforderung für Pharmaunternehmen besteht darin, dass sie jetzt möglicherweise mit Benannten Stellen in Kontakt treten müssen. Dies führt einen neuen Aspekt in die Rolle pharmazeutischer Aufsichtsteams ein. Das Verständnis der Regulierung und dieses Wissen richtig anzuwenden, ist eine Kunst für sich. Die Entwicklung der Konformitätsdokumentation und die anschließende Zusammenarbeit mit Benannten Stellen ist zusätzlich dazu auch zeitintensiv.
Eine weitere Herausforderung besteht darin, dass die Ernennung der Benannten Stellen unter dem MDR nur schleppend vorankommen. Die Kapazitäten der Benannten Stellen, die die Konformitätsbewertung im Rahmen des MDR durchführen, sind jetzt schon ausgelastet. Zum Zeitpunkt der Abfassung dieses Textes sind nur vier Benannte Stellen im Rahmen des EU-MDR ernannt. Bis Ende 2019 werden voraussichtlich ein Dutzend Benannte Stellen beauftragt. Dagegen wurde bisher keine einzige Benannte Stelle im Rahmen der EU-IVDR benannt. Bestandschutz ist nicht zulässig, weshalb alle Hersteller von den Regulierungen betroffen sind, selbst wenn ein Produkt seit vielen Jahren Erfolg auf dem Markt genießen konnte. In diesem Sinne ist es ratsam, dass Hersteller von Kombinationsprodukten so früh wie möglich mit der zuständigen Benannten Stelle interagieren. Die Fristen und Anforderungen für die Einhaltung der Vorschriften sollten angepasst werden, um die Verfahren und die Liaison mit der Benannten Stelle einzubeziehen.
Klinische Daten
Nach dem EU-MDR benötigt jedes einzelne Gerät eine neue CE-Kennzeichnung, um Zugang zum EU-Markt zu gewährleisten. Zur Erzielung der Konformität ist die Einreichung aktualisierter klinischer Bewertungsberichte grundsätzlich. Somit können die Sicherheit, Leistung und Zweck des Medizinprodukts belegt werden. Hersteller, die diesen Schritt zum ersten Mal machen, wird geraten, in Ressourcen zu investieren, damit reibungslose Verfahren standardisiert werden. Außerdem ist es ratsam, herauszufinden, welche Schritte zur Beschaffung fehlender Daten erforderlich sind. Es ist besser, jetzt Zeit und Ressourcen zu investieren, da die klinische Bewertung ein kontinuierlicher Prozess ist. Nur so kann lückenlose Konformität erreicht werden.
Die EU-IVDR und therapiebegleitende Diagnostika
Pharmaunternehmen, die Begleitdiagnostika herstellen, werden ebenfalls einer genaueren behördlichen Kontrolle unterzogen. Therapiebegleitende Diagnostika sind IVD-Geräte, die vor und / oder während der Behandlung angewendet werden, um die Sicherheit und Wirksamkeit eines Arzneimittels zu erhöhen. Die IVDR profitiert von einer längeren Übergangsfrist als die MDR. Hersteller haben Zeit bis zum 26. Mai 2022, um die Anforderungen der neuen Regulierung einzuhalten. Unternehmen, die ihre Begleitdiagnostika in Verkehr bringen oder auf dem Markt behalten möchten, müssen sich auf die Fristen sowohl ihrer Benannten Stelle als auch der nationalen Zertifizierungsstelle für das betreffende Arzneimittel (oder der Europäischen Arzneimittelagentur) verlassen, weil Das IVD-Gerät zusammen mit dem Zielarzneimittel entwickelt werden muss. Es sind bisher keine Benannte Stellen für die IVDR gelistet, daher ist hier ferner mit Verzögerungen zu rechnen. Hersteller sollten sicherstellen, dass sie gut positioniert sind, sobald die zuständigen Benannten Stellen veröffentlicht werden. Aufgrund des neuen Klassifizierungssystems werden viele In-vitro-Produkte in eine höhere Klasse umklassifiziert und werden möglicherweise zum ersten Mal von einer Benannten Stelle überprüft. Ebenso wie beim MDR, ist Bestandschutz nicht gestattet, weshalb für Produkte eine CE-Kennzeichnung notwendig ist, um Zugang zum EU-Markt zu ermöglichen.
Fazit
Unternehmen, die Kombinationsprodukte herstellen, müssen sich bewusst sein, wie sich die EU-MDR auf sie auswirkt, und sollten damit anfangen, die Einhaltung der Frist zu planen. Ansonsten besteht das Risiko eines eingeschränkten Marktzugangs, bis ihnen eine Zertifizierung oder ein Marktzugang erteilt wird. Viele pharmazeutische Aufsichtsteams begeben sich auf Neuland, da sie mit dem Regulierungssystem für Medizinprodukte und der effektiven Zusammenarbeit mit einer Benannten Stelle nicht vertraut sind. Deshalb ist rechtzeitige Planung von entscheidender Bedeutung. Die Zusammenarbeit mit fachkundigen Dritten kann von unschätzbarem Wert sein, um den Prozess zu erleichtern. Es ist wichtig zu bedenken, dass Konformität nicht ein einmaliger Prozess ist, weshalb Unternehmen Einhaltungsverfahren in ihre Abläufe integrieren sollten, damit sie für zukünftige Überprüfungen gut aufgestellt sind. Proaktive Unternehmen, die eine Weitsicht haben, sind gut vorbereitet, um eine nachhaltige Einhaltung gesetzlicher Vorschriften zu erreichen, die ihr Geschäft letztlich vorantreibt.
Autorin: Elizma Parry – Director, Global Clinical Practice bei Maetrics